
Inyectan ADN editado en un paciente para curarle una enfermedad mortal
CientÃficos de EEUU completaron un nuevo tratamiento en un paciente con una enfermedad mortal y buscan modificar su genoma humano con la int
Un nuevo hito en la historia de la ciencia ha tenido lugar estos dÃas en Estados Unidos. Un grupo de investigadores del UCSF Benioff Children's Hospital en Oakland completó un nuevo tratamiento en un paciente con una enfermedad mortal y buscan modificar su genoma humano con la intención de curar su trastorno genético.
Se trata del primer procedimiento de edición de un gen dentro de un cuerpo humano vivo. Desde hace varios años, cientÃficos en todo el mundo vienen trabajando en varios niveles dentro de la terapia genética, donde buscan modificar genes en un laboratorio antes de volverlos a incorporar a un ser humano.
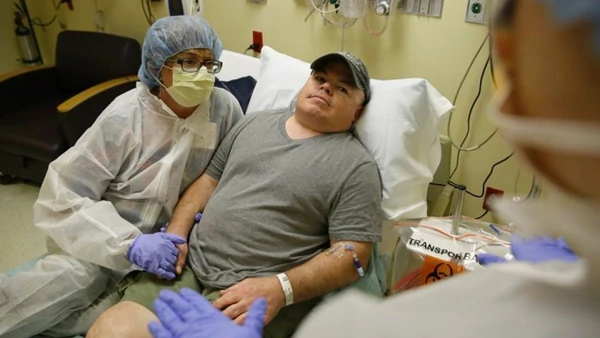
Por ello, el avance cientÃfico realizado en el paciente estadounidense Brian Madeux, de 44 años, podrÃa cambiar para siempre el tratamiento genético de padecimientos que hasta ahora son incurables.
Madeux, que sufre un trastorno metabólico raro llamado SÃndrome de Hunter, recibió un tipo de medicamento vÃa intravenosa, el cual contiene miles de millones de copias de un gen corrector que está unido a una herramienta genética que es capaz de modificar su ADN.
Esta herramienta es conocida como 'Zinc Finger Nucleases' y es anterior a CRISPR. Su trabajo consiste en cortar el ADN en el lugar correcto, como si fuese una "tijera molecular", eliminar el gen responsable del padecimiento e insertar el gen correctivo. Las instrucciones de todo este complejo procedimiento están codificadas en un virus que fue modificado para dirigirse al hÃgado del paciente.
Un caso inédito
El SÃndrome de Hunter es causado por una enzima faltante o en mal funcionamiento. Esto ocasiona que el cuerpo de Madeux no pueda descomponer ciertos carbohidratos, que luego se acumulan y causan daño. Es problema puede producir un aumento en el tamaño de la cabeza y el hÃgado, pérdida de la audición, rigidez de las articulaciones, dificultad para respirar e incluso problemas mentales. Lamentablemente, a dÃa de hoy no existe una cura y su tratamiento consiste en terapias enzimáticas cada semana.
"¡Este es el año de la genética!", se entusiasma en declarar el genetista argentino Jorge Dotto. Y su exclamación tiene mucho sentido.
Primero surgió la noticia de los cientÃficos Jeffrey Hall, Michael Rosbash y Michael Young, que ganaron el Premio Nobel de Medicina 2017 por sus descubrimientos sobre los mecanismos moleculares que controlan nuestros relojes biológicos. Luego se conoció que un equipo de investigadores chinos consiguió corregir una anomalÃa genética en embriones humanos, portadores de una grave enfermedad hereditaria de la sangre.
Y por último, la Agencia Federal de Alimentos y Medicamentos de Estados Unidos (FDA) aprobó hace pocos meses la comercialización de la primera terapia génica en el mundo, que consiste en modificar genéticamente el sistema inmunológico de un enfermo para combatir una agresiva forma de leucemia.
"Y ahora esto. Es una noticia increÃble. Estamos en el camino de la medicina personalizada. Y se trata de un momento histórico, que por supuesto debemos tomar con cautela y responsabilidad, ya que el campo de la medicina genética es muy amplio y falta mucho todavÃa que conocer", agregó el especialista.
"Es una muy buena noticia, ya que durante tres horas se le inyectó a un paciente con sÃndrome de Hunter un medicamento que utiliza una tijera genética en el propio cuerpo, donde actúan las moléculas del ADN", precisó Dotto.
Esta enfermedad, conocida como "el Alzheimer de los bebés", pasa inadvertida en la revisión ordinaria de un recién nacido y se comienza a manifestar en los primeros años para acelerar su desarrollo de sÃntomas y señales a medida que pasa el tiempo.
"Las personas que nacen con el SÃndrome de Hunter no pueden metabolizar una clase de azúcares que contribuye al desarrollo de los huesos, la piel y los tendones, entre otros tejidos; esos azúcares se acumulan en las células y van dañando diferentes partes del organismo, incluÃdo el cerebro", precisó Dotto.
La imposibilidad de procesar los mucopolisacáridos se debe a la falta de una enzima, por lo cual la enfermedad que el médico escocés Charles Hunter describió por primera vez en 1917 se conoce también como Deficiencia de Iduronato Sulfatasa y Mucopolisacaridosis Tipo II (MPS II).
Algún dÃa, los investigadores podrán usar la edición de genes para reparar el gen defectuoso en las células que causa enfermedades como el sÃndrome de Hunter. Sin embargo, ese no es el objetivo del ensayo, patrocinado por Sangamo Therapeutics, una compañÃa de biotecnologÃa con sede en Richmond, California.
En cambio, según datos de la compañÃa, los médicos insertaron una copia de reemplazo del gen, usando la edición de genes para cortar la hélice de ADN de las células hepáticas en un lugar especÃfico cerca del promotor, o el interruptor de encendido y apagado, para el gen de una proteÃna llamada albúmina.
Las células corrigen el daño insertando el ADN del nuevo gen, suministrado por los investigadores junto con las tijeras de ADN del editor genético, y la actividad del gen luego es controlada por el potente promotor de la albúmina. La idea es convertir estas células hepáticas modificadas en una fábrica para hacer que la enzima desaparezca en el sÃndrome de Hunter.
El enfoque especÃfico de Sangamo, conocido como "puerto seguro", deberÃa evitar los riesgos de utilizar la terapia génica tradicional para alterar el genoma de una célula, que pega en el nuevo gen en un lugar aleatorio en el genoma y potencialmente puede activar un gen canceroso. Y debido a que el cuerpo no necesita mucha de la enzima, modificar solo una pequeña fracción de las células del hÃgado deberÃa ser suficiente para tratar la enfermedad.



